产品
FIB-SEM
Nanomanipulators
OmniProbeOmniProbe Cryo软件
AZtec3DAZtecFeatureAZtec LayerProbeTEM
Hardware
EDSUltim MaxXploreImaging
软件
AZtecTEM
 牛津仪器集团成员
牛津仪器集团成员

15th July 2020 | Author: Anthony Hyde
The Pharmaceutical industry is one of the most regulated industries in the world and carrying out SEM-EDS analysis within such a regulated environment can be problematic.
The FDA in the USA is responsible for two of the most common sets of regulations or guidelines. The first is 21 CFR Part 11. This regulation covers the use of electronic records and electronic signatures in the regulated pharmaceutical, biotechnology and medical device industries.
It is intended to ensure that electronic records are trustworthy and reliable. The rules require computer systems and applications to have; adequate security features - audit trails to automatically monitor creation, modification and deletion of electronic records - unique electronic signatures - working backup and recovery and disaster recovery procedures - effective and rapid archive and restore mechanisms.
The second is a set of guidelines called GxP which were created to ensure that products are safe and meet their intended use. They are applicable to products that could potentially put peoples’ safety at risk i.e. in industries such as pharmaceutical, food, medical device, and life sciences. GxP is a general abbreviation for the "Good Practice" and the "x" indicates a particular field. Some common examples are:
The central aspects of GxP focus on Traceability and Accountability through Documentation, i.e. the ability to see the development of a process or analysis and who contributed and when.
Although these are US regulations and guidelines, their impact is felt strongly outside the US. International companies who wish to sell regulated products into the US must adhere to these rules and guidelines. Registered organisations can be audited by the FDA. In the US, organisations that fail audits can be heavily fined or even closed. Organisations outside the US can be prevented from exporting to the US.
There is a European equivalent of 21 CFR Part 11, which is called ‘EU GMP Annex 11’. These guidelines are very similar to 21 CFR Part 11, but Annex 11 is not a regulation.
Analysts in the pharmaceutical industry must also to be mindful of IQ, OQ and PQ.
IQ, Installation Qualification, is essentially a proof that an instrument has been installed correctly. OQ, Operational Qualification, and PQ, Performance Qualification, look at proving that the instrument meets specification and operates as it was designed to.
Because laboratories in these regulated environments will have numerous analytical systems with varying levels of compliancy, they must create Standard Operating Procedures (SOPs) for each system and analysis carried out in their laboratory. These SOPs will not only help users follow the same process consistently, but they will also help prove compliancy when audited (by either a customer or an FDA officer). So, analysts look to instrument manufacturers to help make these SOPs as simple and straightforward as possible.
To deliver an EDS solution for these regulated environments we worked with users from around the world. This gave us an insight not only into what was required from our software from a regulatory viewpoint, but also from a day to day usability viewpoint i.e. any software solution must satisfy the core regulatory requirements of data security, traceability, accountability and integrity, but not at the expense of usability.
From this, we created AZtecPharma, which is a dedicated version of AZtecLive with all the accuracy and reliability, but with enhanced data security and integrity. It enables EDS analysis to be incorporated into laboratory SOPs to support good practice quality guideline ‘GxP’ and for regulations such as 21 CFR part 11, EU GMP Annex 11, and also helps with IQ, OQ and PQ. The software includes all the tools required to help users quickly analyse and characterise their sample with confidence and ease. Tools such as ‘Spectrum Acquisition’, ‘X-ray Mapping’ and ‘LineScanning’ help determine which elements are present in a sample and how they are distributed, but in addition, it has Increased Security, Digital Signatures, Audit Trails and an Inspector program to monitor the Audit Trail.
One of the users involved in the development of AZtecPharma was Jill Webb from Reading Scientific Services. RSSL are a world-leading Contract Research Organisation based in the UK. They provide analytical testing, R&D, consultancy and training, mainly to the food and pharmaceutical sectors. They have a range of equipment from FT-IR to SEM-EDS, each of which may not fully comply with GxP and CFR requirements. They perform a ‘Gap’ and Risk assessment on each of the instruments in their laboratory to determine any areas where data integrity may be an issue. An SOP is then created to detail how the instrument should be used and how data is saved.
AZtecPharma uses the local windows credentials, which makes it easy for the local IT department to set up and control all aspects of user access to the AZtecPharma system. This includes who can login to the computer, automatic logging out when inactive and password rules.
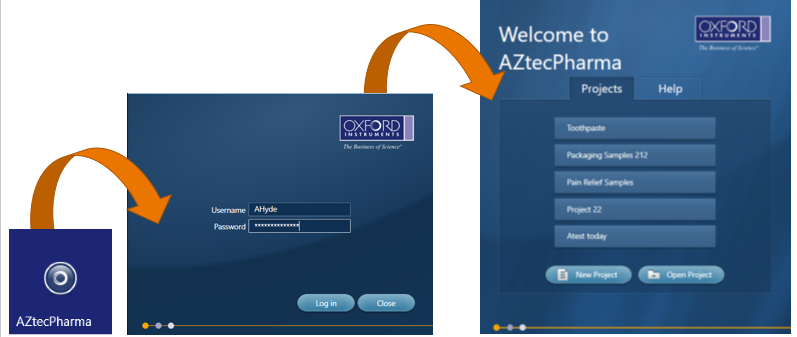
Once the user is logged into AZtecPharma, there is little difference to the operation of the standard AZtecLive software, apart from the inability to delete data from the data tree. When an analysis is being carried out, AZtecPharma records when an action has been carried out and by who and creates an audit trail that is saved in a secure database.
To view this ‘Audit Trail’ we have a separate program called ‘Inspector’, which is opened in the same manner as the AZtecPharma software.
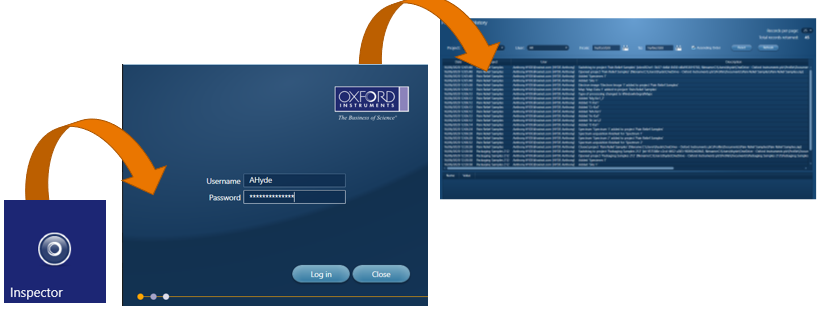
Users can filter records based on project name, username or date/time. Any Spectrum, Map or LineScan acquisition will include the acquisition details in a separate window. The records in this audit trail cannot be modified or deleted.
To hear directly from Jill Webb and myself about how AZtecPharma has helped RSSL and others to comply with 21 CFR Part 11 and EU GMP Annex 11, watch our recent webinar on Operating SEM-EDS in a regulated environment.
We send out monthly newsletters keeping you up to date with our latest developments such as webinars, new application notes and product updates.

© 牛津仪器 2026
 公安机关备案号31010402003473
公安机关备案号31010402003473
